Hormonal therapy in breast cancer

Breast cancer still remains one of the most causes of death worldwide among women patients. The incidence rate is still high in Europe and each year 350,000 new cases are diagnosed and 13,000 death cases are registered. It has been documented that predisposing risk factors such as age, environmental, lifestyle, genetics, and reproductive factors may be counted strongly for breast cancer initiation. Different strategies have been proposed for breast cancer therapy. Endocrine breast cancer treatment holds a great future promise in terms of controlling breast cancer and reduces mortality. Emerging pharmaceutical compounds have shown high efficacy against breast cancer. However, none of them have been straightforward successful in treating breast cancer because of drug resistance. In this review, I will shed the light on the most common endocrinal therapeutic drugs used in breast cancer treatment and their pharmacological points of view and how resistance to drugs opens a new era in assisting breast cancer therapy. Over 70% of breast cancer can be classified histopathologically into two main types; in situ forms in which cancer has not invaded to the basement membrane and the invasive form which spread not only through the basement membrane but also to other vital organs in the body (Malhotra, Zhao et al. 2010). Morphological classification still used to diagnose and evaluate cancer progression, however, in some heterogenic nature of cancers such as breast cancer this might be insufficient for making an accurate decision for therapy and prognosis since some patients could respond to specific kind of therapy while others not although they apparently have the same classified type of breast cancer. A more recent advanced classification based on molecular and immunohistochemical investigations revealed new molecular markers such as hormone receptors estrogen (ER)/ Progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER-2) which can determine the clinical course of the disease and can be utilized pharmacologically to get effective therapeutic outcomes (Rakha, Reis-Filho et al. 2010). For example, it has been found that blocking hormonal receptors was among the best effective treatment of hormone-responsive breast cancer patients and has the least toxic side effects (Pearce and Jordan 2004) thus raising the fact that endocrinal manipulation in breast cancer therapy could be a powerful tool to combat cancer. The discovery of selective estrogen receptor modulators, SERMs, such as Tamoxifen and its derivatives has revolutionized cancer treatment strategies and become the anti-hormonal drug of choice for breast cancer. In this review, we will discuss some pathophysiological and molecular aspects of anti-hormonal agents used against breast cancer and we will approach their pharmacological roles based on literature findings.
Molecular structure of the Estrogen receptor
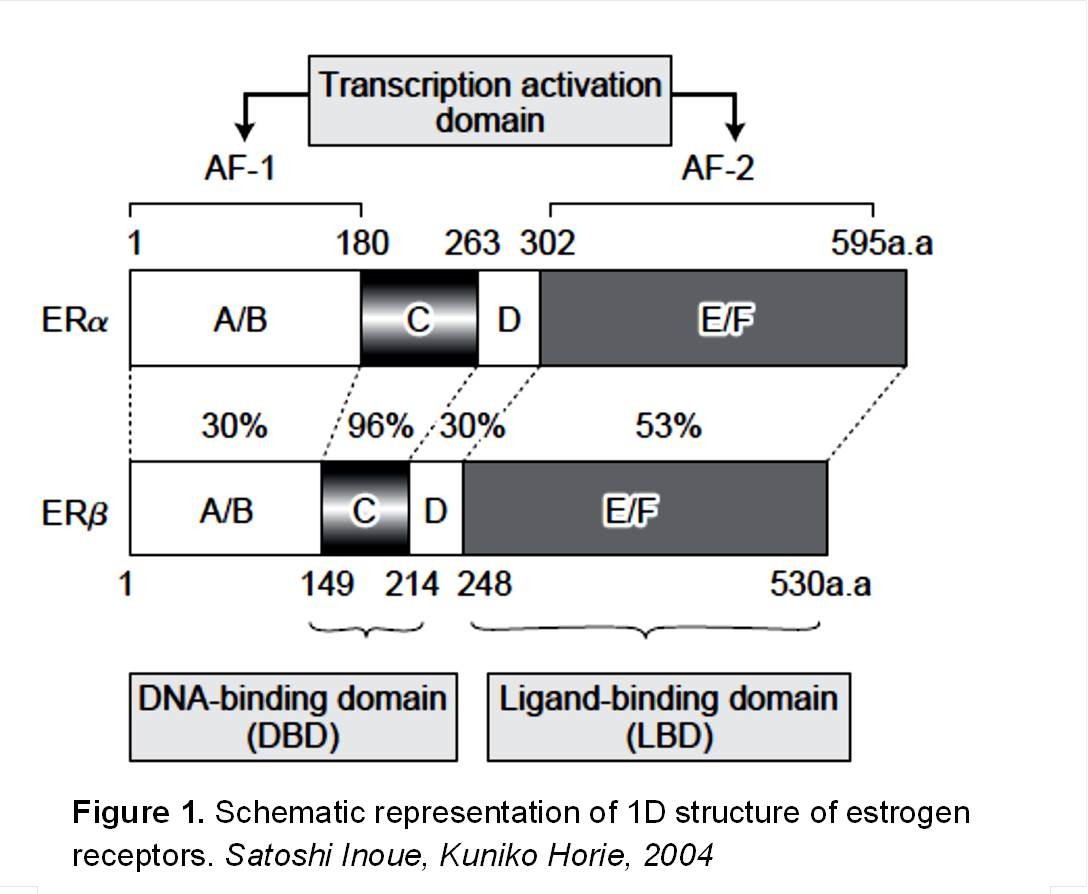
Estrogen receptors are of two types; alpha and beta. They are nuclear receptors of steroid/thyroid hormone superfamily and they consist of three independent functional domains (Figure 1); NH2 –terminal or A/B domain, DNA-binding domain, and the COOH-terminal or ligand-binding domain (Nilsson, Makela et al. 2001). The amino-terminal domain encodes for a ligand activation function called AF1 which is responsible for protein-protein interactions and transcription activation. DNA binding domain binds specific DNA sequences and has a role in receptor dimerization. The carboxyl-terminal promotes nuclear translocation, receptor dimerization and mediates ligand binding (Hewitt and Korach 2002).
Physiological roles of Estrogen Receptors
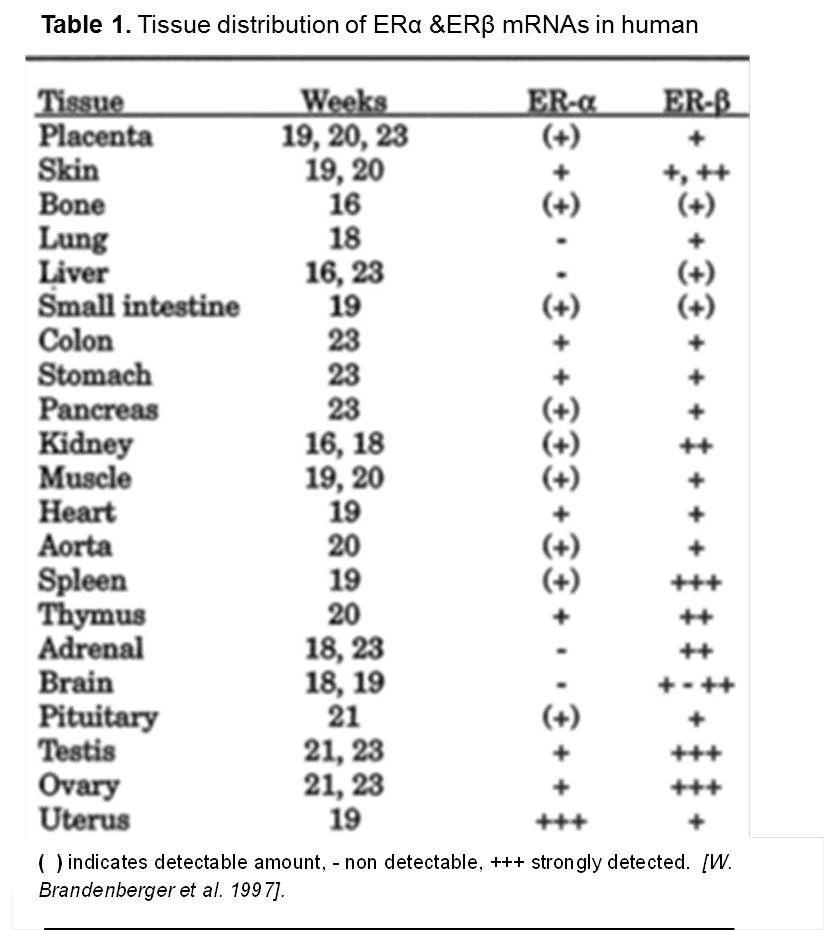
In a fascinating highly cited old study, Alfred. W. Brandenberger et al was able to demonstrate different tissue distribution of estrogen receptors, alpha and beta as summarized in (Table 1.) The High mRNA expression pattern reflects the fact that these tissues are more affected by ER ligands than the others. More methods have been developed to investigate the physiological role of ERs and their impact on different tissues most important ones were those applied to estrogen receptors knockdown mice models. Knockdown ERs in male and female mice revealed a different physiological role of ERs gene functions, however, neither ERα nor ERβ knock-down was lethal to the mice (Pearce and Jordan 2004).
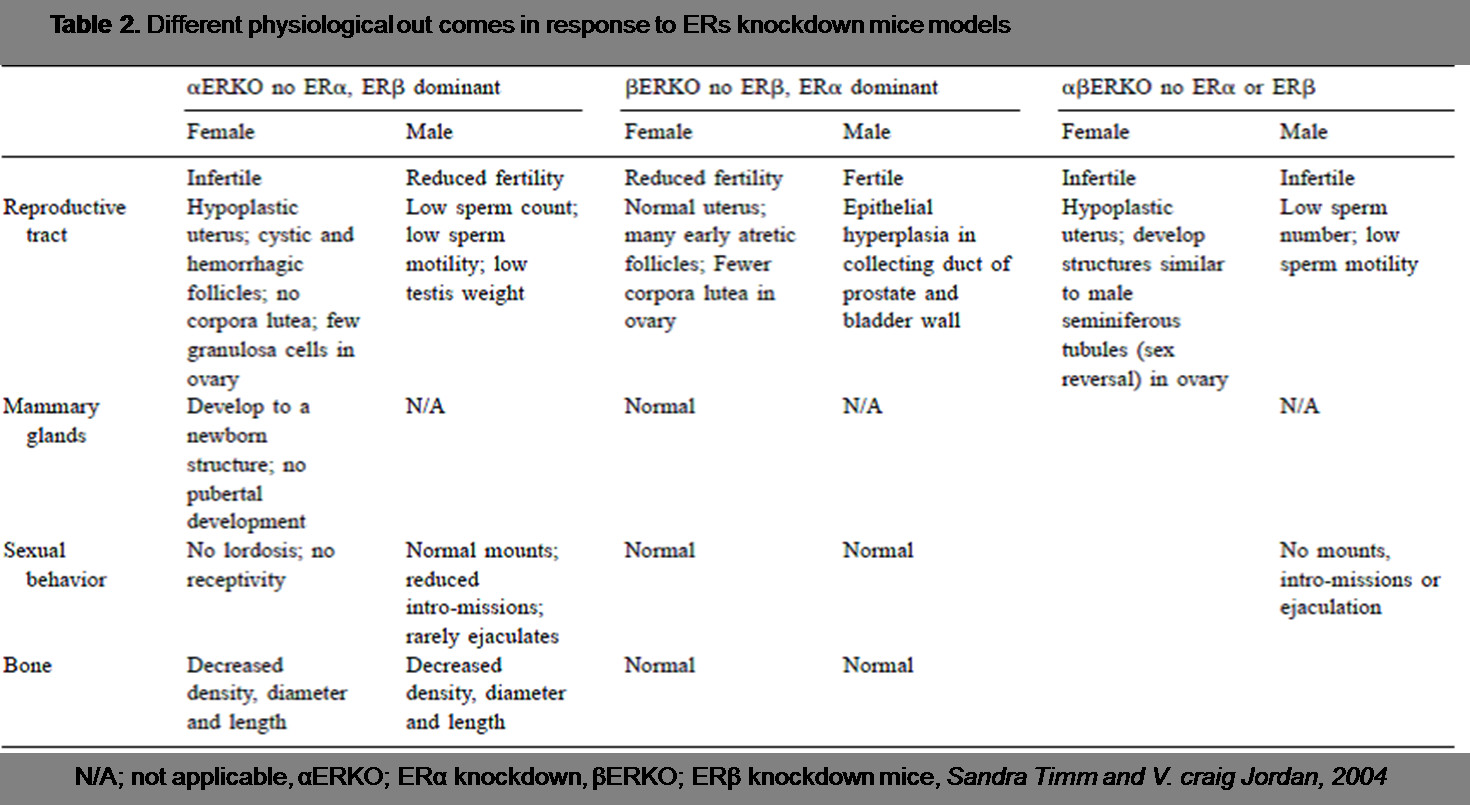
(Table 2.) highlights the most important physiological findings as a result of ERs knockdown models.
Roles of Estrogen Receptors in breast cancer
Accumulative evidence based on animal models studies indicated that estrogen increases the incidence of cancer in mammary glands, vagina, uterus, and testis (Pandey, Pandey et al. 2011). Other lines of evidence-based on epidemiological studies shown that administration of estrogen-containing oral contraceptives or increased serum levels of estrogen were associated with increased risk of breast cancer (Fuhrman, Brinton et al. 2012). It has been reported that estrogens have an important role in the development and growth of breast cancer (Platet, Cathiard et al. 2004). Young-Rae Lee et al. showed that stimulation of two breast cancer cell lines, MCF-7, and MDA-MB-231 by 17β-estradiol (E2), the most potent estrogen, increases breast cancer cell proliferation (Lee, Park et al. 2005). Moreover, it is worthy to note that only MCF-7 cancer cell line showed increased breast cancer growth. The two distinct both estrogen receptors, ERα and ERβ, while the other cell line was expressing only ERβ and did not respond to estrogen treatment (Lee, Park et al. 2005). Deep investigations in the signaling pathways also revealed that increased activity of AKT and PIP3 leads to increase cell proliferation induced by estrogen (Jiang and Liu 2009). Other constructive works have been done to the target aromatase enzyme, a key enzyme in estrogen biosynthesis, which inhibits the production of estrogen and results in breast cancer regression (Brueggemeier, Hackett et al. 2005). Taken together, these findings indicate that targeting these molecular targets could be a novel approach for breast cancer hormonal therapy.
Hormonal therapy of breast cancer
Antiestrogens (SERMs) Selective estrogen receptor modulators
(SERMs) are believed to have multiple activities. They antagonize estrogen receptors in some tissues such as the uterus and mammary glands while they agonize others like those in bones and cardiovascular tissue. Such effects have been demonstrated to have osteo and cardio- preventive roles in postmenopausal women (Albertazzi and Purdie 2001) however, serious side effects including blot clots, and endometrial cancer, have been reported with the use of SERMs (Maximov, Lewis-Wambi et al. 2009). SERMs include toremifene, raloxifene, and fulvestrant, and the most well-known SERMs, Tamoxifen.
Tamoxifen
Pharmacology of Tamoxifen
Since the discovery of antiestrogenic, Tamoxifen, role in breast cancer and it becomes the anti-hormonal drug of choice in positive estrogen receptors breast cancer (Stendahl, Ryden et al. 2006). Tamoxifen mediates its effect by blocking estrogen receptor through competing with its natural ligand thereby inhibits the transcription of genes regulated by estrogen receptor elements, EREs, involved in activation of cell proliferation and programmed cell death (Mandlekar, Hebbar et al. 2000). Tamoxifen is a triphenylethylene and exists into two main isomers; cis and trans tamoxifen (Osborne, Wiebe et al. 1992). The trans isomer has a higher affinity for the estrogen receptor than cis tamoxifen. Tamoxifen itself is a pro-drug and when metabolized in the liver it liberates N-desmethyltamoxifen and trans-4-hydroxytamoxifen which are very potent estrogen receptor blockers (Swaby, Sharma et al. 2007). After oral administration, Tamoxifen and its metabolites are readily absorbed with serum half-lives ranging from 7 – 14 days of 20 mg/day dosage. A study of animal rat breast cancer models showed that Tamoxifen has a lower half-life than those of its metabolites (Mandlekar, Hebbar et al. 2000). Tamoxifen serum concentration has declined rapidly after s.c administration to the baseline within 5 hours while trans-4-hydroxytamoxifen metabolite reaches Cmax of 385 ±132 ng/ml at 5 h and declined to the baseline within 7 hours. Moreover, N-desmethyltamoxifen found to have a higher Cmax of 2136 ± 1516 ng/ml at 48 h, and goes back slowly to the baseline within 96 hours. The study also showed that the total serum exposure in rats for Tamoxifen was very low compared with its metabolites by measuring the area under serum concentration curves AUC. These metabolites were found also to have higher concentrations in tissues more than the elimination phase of Tamoxifen metabolites (Clarke, Leonessa et al. 2001). It is of importance to note that these pharmacodynamics observations could be also related to the nature of substrate specificity of both Tamoxifen metabolites and to the fact that estrogen receptors expression is higher in breast cancer tissue than other tissues (Mandlekar, Hebbar et al. 2000) indicating higher Tamoxifen concentrations in tissues than in serum. This higher intratumoral concentration of antiestrogens over estrogens can explain at least in part the effective therapeutic response in ER-positive patients of breast cancer. In a long-term therapy profile, Tamoxifen metabolites may reach stable and steady-state serum levels for up to years (Osborne 1998) and this is of advantage especially in cases required a long duration of Tamoxifen treatment such as in breast cancer (Swaby, Sharma et al. 2007). In a recently published study, a large randomized trial on women of early breast cancer has indicated that Tamoxifen increases survival rate and reduce recurrence in those patients being on treatment for 5 years compared to those for 2 years of tamoxifen therapy (Thomssena and Harbeckb 2010).
Aromatase inhibitors (AIs)
Pharmacology of Aromatase inhibitor
Aromatase is a key enzyme catalyzes the rate-limiting step conversion of androgens to estrogens (Bhatnagar 2007). Aromatase and estrogens are produced by functional ovaries and many other tissues such as breast, fat, connective tissue and bones (Smith and Dowsett 2003). They can also be produced by cancer tissues despite the patient is post or premenopausal. While Tamoxifen considers the antiestrogenic drug of choice in positive ER-breast cancer patients, aromatase inhibitors are the second-line drug of choice in premenopausal breast cancer and the first-line drug in combined adjuvant therapy in postmenopausal patients (Fabian 2007). Aromatase inhibitors are divided into two main groups; Type 1 (steroidal) and Type 2 (non-steroidal) inhibitors (Narashimamurthy, Rao et al. 2004). Steroidal inhibitors type is known also as irreversible inhibitors as they bind strongly to the enzyme and become reactive intermediates called aromatase “inactivators” which eventually shut down the enzyme activity. Non-steroidal type binds reversibly to the heme prosthetic group of the enzyme and prevents binding with its natural substrate by competitive inhibition (Johnston and Dowsett 2003). AIs also can be classified according to their consecutive development into three generations (Figure 2.) (Hong, Didwania et al. 2009). It has been proved that all aromatase inhibitor generations are well tolerated and much more potent than Tamoxifen with minimum toxic side effects in breast cancer treatment (Chumsri, Howes et al. 2011). Currently, AIs first and second generations are no longer in clinical use because of their adverse effects regardless of their high efficacy in combating the metastatic type of breast cancer. The need for such high aromatase inhibitors with low toxic outcomes led to tremendous efforts to develop new compounds with improved toxicity profiles. These efforts have been successful in obtaining the third AIs generation such as lertozole, anastrolzole, and exemestane and they showed superior effect breast cancer therapy compared with the second generation which showed the efficacy of 90% aromatase inhibition (Geisler, Haynes et al. 2002). Table 3. Summarizes the pharmacokinetics of the most common third AIs generation of clinical importance.

Anti-hormonal therapy resistance
Around 30% of advanced ER-positive breast cancer patients will no longer respond to AIs or Tamoxifen therapy because of de novo resistance or resistance developed over time after initial therapy, acquired resistance (Riggins, Schrecengost et al. 2007). Several mechanisms have been reported to date in different in vivo and in vitro studies including upregulation of HER-2 and ER due to very low levels of estrogens, increase endogenous estrogens, upregulation of molecular signaling factors such as AKT and PIP3. Other pathways associated with insulin growth factor and EGFR, ERK/MAPkinases also have been documented (Gutierrez, Detre et al. 2005). Recently, it has been widely accepted that understanding the molecular mechanisms behind cross-talking between molecular signaling pathways is a key step to develop new agents targeting these molecular growth factors and receptors themselves. It is well known currently that intracellular kinases play a major role in vital cell functions such as proliferation, differentiation, migration and cell survival and targeting these enzymes can block or reverse resistance to anti-hormonal breast cancer therapy such as trastuzumab (Herceptin) which targets HER-2 and ABX monoclonal antibodies against EGFR (Jones 2003).
Summary
Endocrinal breast cancer therapy has elucidated the role of estrogen receptors in breast cancer and its signaling pathways by which cancer cells proliferate and grow. Interestingly, Tamoxifen and aromatase inhibitors have a great impact on breast cancer regression and both could differ in their pharmacology and pharmacokinetics but they still widely used based on the biological nature and molecular classification of the cancer tissue. Resistance to anti-hormonal breast cancer treatment revealed some pivotal molecular mechanisms which could be of great interest for developing new efficient compounds targeting cellular and molecular targets at once with the highest efficacy and the lowest side effects. Several molecular targets have been identified and require fundamental work to be characterized before introducing them to the market.
c3ckWavzlDw
0MsIDfhICd0
294m38o3dC7
kDPpnFiJcx5
cOZUXOoM3Fu
WfnXQVpfDCn
h4YQDzzXElS
DKdS2DuJhgV
Dq2w3oqiT9j
Pb8J1kI6CJB
jihAkC0tw5J
Ix1imUT4QvE
qdGEIHwdsfo
x0F82nKPl2H
wF0HySEwKJM
xLddGRWg9eO
DxcmbbHrJRi
pk4MOIFkIic
OZVEbjkG2EX
yaFzCMWLDFj
kFWYMXEA81D
OwOhS1Zz7ce
vyNRPlHTudV
N1VuB9h6Ipl
WudO1lPwduQ
SKe27Ws3rXw
D7CSDwh6yMt
DzG96SOQzlT
gFnaBsllFjo
qJTyXnJLX53
5dySi2SNqsH
t2S0hQcZowe
KwZmAIkxGEJ
qgqVAar0rhN
OoIVrpG1Dfs
tujlo7KbSgy